I want to share with you the new section of Didactic Materials at the Sixth Researcher website.
In this section I will include courses, presentations, workshops and other materials that I prepared and could be useful for other researchers.
At the moment you can download 10 lessons with the fundamentals of Python 3 for biologists and other scientists that I imparted at UAM.
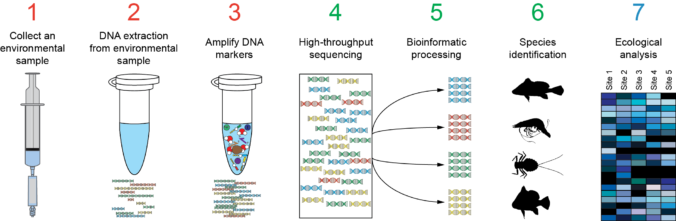
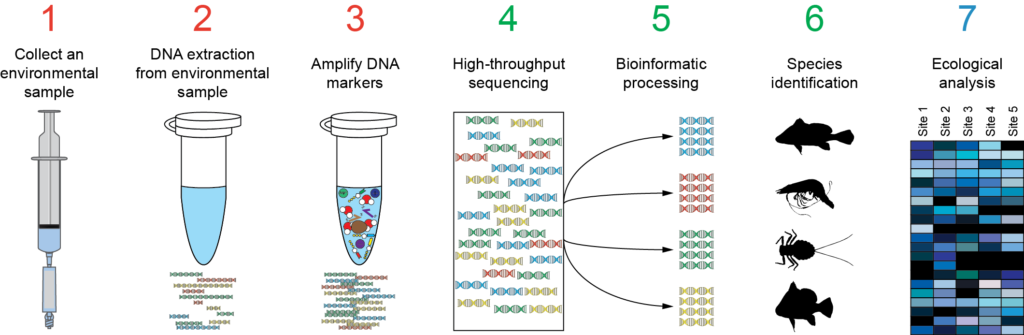
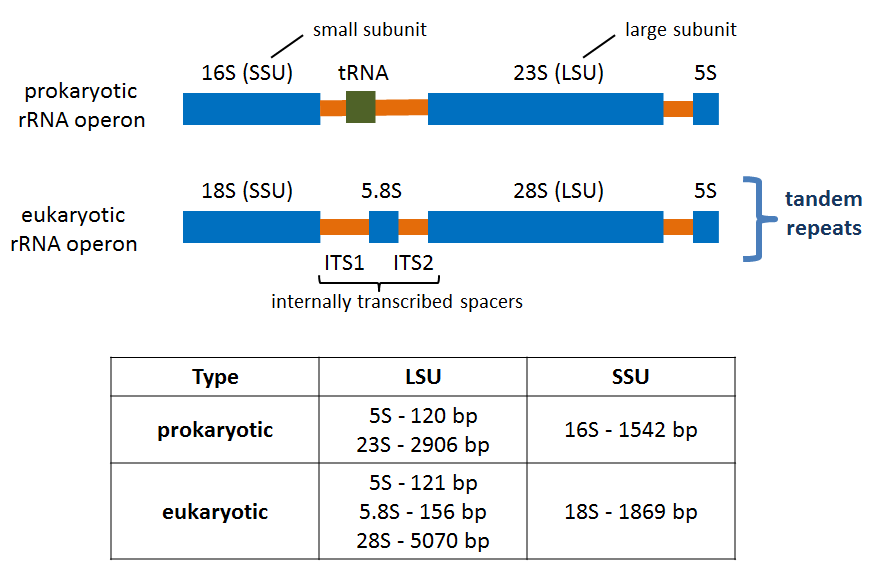
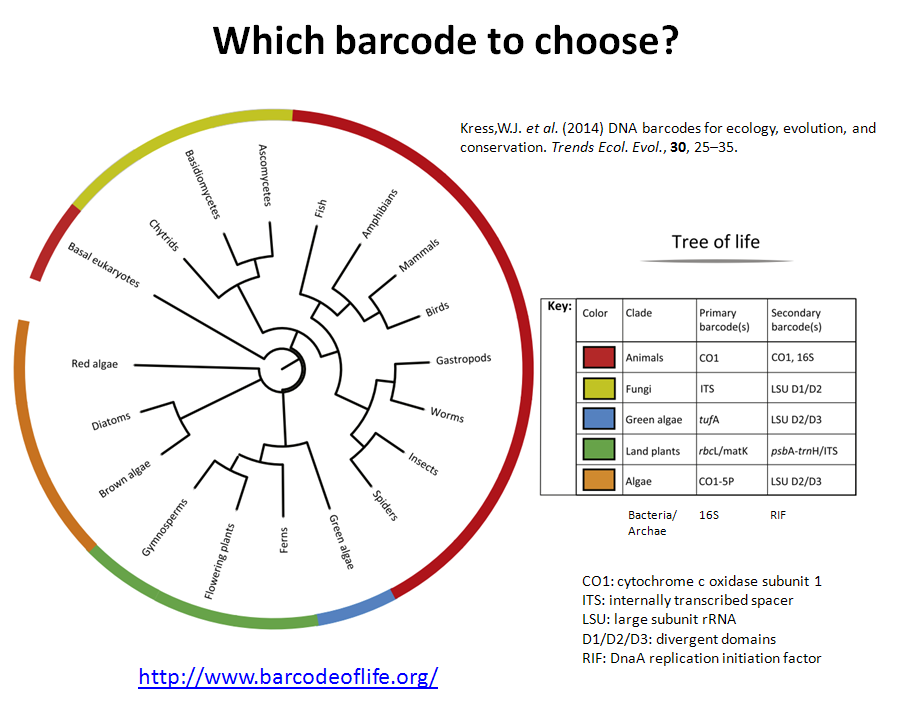
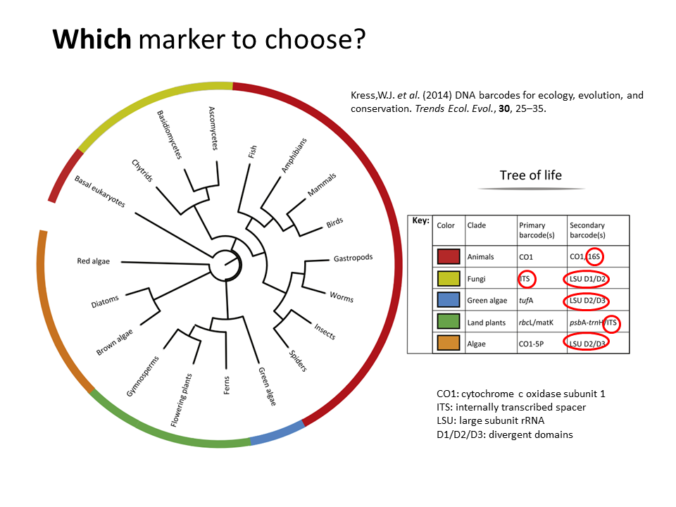
Also is available a Metabarcoding workshop with the fundamentals of the technique and a practical example explaining the bioinformatics analysis of the data.
 The didactic materials in this new section will be licensed as Creative Commons Attribution-NonCommercial.
The didactic materials in this new section will be licensed as Creative Commons Attribution-NonCommercial.
Python for Scientists
Materials from the course Basic Python 3 Programming for Scientists imparted at Adam Mickiewicz University:
- Python3 reference_cheat_sheet for beginners.
- Lesson 1. Running Python 3 in Windows.
- Lesson 2. Data structures: lists and dictionaries.
- Lesson 3. Data comparison and conditional statements.
- Lesson 4. User data input and ‘while’ loops.
- Lesson 5. ‘for’ loops and functions.
- Lesson 6. Built-in functions. Reading and writing files.
- Lesson 7. Python, Spyder, Jupyter and Anaconda.
- Lesson 8. Python scripts and modules.
- Lesson 9. NumPy, SciPy and Matplotlib.
- Lesson 10. Biopython.
Metabarcoding Bioinformatics analysis
Materials from the workshop Introduction to Bioinformatics analysis of Metabarcoding data: